Research Article - (2014) Volume 3, Issue 2
Fabs fragments (Fabs) maintain binding ability to specific antigens, but lack the binding site for complements and for receptors on leukocytes that play a crucial role in Rheumatoid Arthritis (RA). In the present study, we investigated whether type II Collagen (CII)-Induced Arthritis (CIA) in mice was suppressed by anti-CII Fabs prepared by papain digestion of anti-CII antibodies. CIA was induced in DBA/1J mice by immunization with chicken CII and completes Freund’s adjuvant. To investigate the effect of anti-CII Fabs on the CIA model, mice were injected intraperitoneally with anti-CII Fabs 1 day before the first immunization. As a result, CIA was markedly inhibited by anti-CII Fabs; furthermore, the histological features of anti-CII Fabs-untreated mice included severe hyperplatic synnovium, cartilage and joint destruction, and leukocytic infiltration, whereas animals given anti-CII Fabs showed significant reduction of these histological changes. Additionally, antigen-specific suppression of CIA by anti-CII Fabs was related to the reduced levels of complement C3a in serum. Meanwhile, in vitro studies revealed that anti-CII Fabs significantly blocked the binding of intact anti-CII antibodies and also decreased complement activation. Collectively, the development of CIA was suppressed by anti-CII Fabs, which prevented the binding of anti-CII antibodies to antigen followed by the inhibition of complement activation, suggesting that autoimmune arthritic diseases such as RA may be specifically treated with pathogenic antibody Fabs.
Keywords: Fab fragments, Collagen-induced arthritis, Rheumatoid arthritis, Complement C3a, Mice
Autoantibodies against type II collagen (CII) are well documented in patients with Rheumatoid Arthritis (RA) [1,2]. After multimeric immune complex (ICs) of CII and anti-CII autoantibodies form on the articular cartilage surface, both complement components and FcγR-bearing cells are primarily activated by capturing the Fragment Crystallizable (Fc) portion of cartilage-bound antibodies [3]. The provoked levels of complement-activated products, including C3a and C5a in serum and synovial fluid in RA patient, have been described previously [4,5]. Therefore, the diapedesis of inflammatory cells was induced by chemotactic activity of complement-cleavaged proteins in synovial tissues [6,7]. The infiltrated leukocytes are able to enhance the articular inflammatory response and cause synovial injury by releasing pro-inflammatory cytokines [8]. Moreover, the differentiation of Antigen-Presenting Cells (APC) is promoted by FcR-binding ICs. The immunological reaction is also amplified by highly self-antigen presentation of activated APC to adaptive immune cells [9]. Thus, the Fc portion of autoantibodies is crucial for autoimmune diseases, and blockade of the immuno-pathological action of autoantibody-mediated RA may be an alternative attractive remedy [3]. Fab fragments (Fabs) are prepared by digestion with papain [10]. The structure of the Fab fragment retains monovalent antigen binding sites of their antibodies without the Fc portion [11]. Fab fragments not only have high ability to bind antigenic epitopes but are also lacking the binding site for complement proteins and inflammatory cells. Therefore, therapies using Fab fragments are expected for antibody-mediated diseases by down-regulating the complement pathways and the pro-inflammatory cytokines released from inflammatory cells such as macrophages. The suppression of bullous pemphigoid, autoantibody-induced blistering disease, in mice by recombinant Fabs against the non-collagenous 16th-A domain of type XVII collagen has been recently documented [12]; additionally, ovalbumin (OVA)-specific Fabs has been readily available for Anti-OVA Antibody-Mediated Arthritis (AOA-MA) in mice [13]. However, in murine Collagen-Anduced Arthritis (CIA), which resembles human RA, the inhibitory effect of Fabs has never been examined. In the marketplace, many different products for RA therapies, including steroids, non-steroidal anti-inflammatory drugs, immunosuppressants, and biological agents, are available; however, several chemical agents have shown a risk of adverse effects [14-16]. Unfortunately, antigen-specific therapies have not been clinically used for this disease. In this study, to develop a new therapeutic strategy for the treatment of RA by antigen-specific suppression, we investigated whether Fabs prepared by enzymatic papain digestion from polyclonal antibodies (pAb) against mouse CII inhibits CIA.
Animals
Male DBA/1j mice, 8 weeks of age, were kept in a specific pathogen-free environment of the animal breeding unit. Healthy mice were maintained in a temperature-controlled cage with free access to standard rodent chow and water. All animal experiments were performed in accordance with protocols approved by the Ethics Committee of Kobe Pharmaceutical University, Kobe, Japan.
Induction of arthritis
DBA/1j mice were subcutaneously immunized with 0.1 ml antigen emulsion (50 mg chicken CII (Sigma Aldrich, St Louis, MO, USA) dissolved in 1 mg of 0.1 N acetic acid and emulsified with an equal volume of complete Freund’s adjuvant (CFA) (Difco Laboratories, Detroit, MI, USA)) into the base of the tail on day 0 and 21 [17]. The non-sensitized group was mice receiving no chicken CII immunization. In this study, we focused on the prophylactic intervention of anti- CII Fabs in collagen-induced arthritis; therefore, anti-CII Fabs (1 mg/mouse) was intraperitoneally administered 1 day before the first immunization. Control mice were given the same amount of normal IgG Fabs (Rockland Immunochemicals, Gilbertsville, PA, USA).
The mice were observed for clinical arthritis every other day after the second immunization with chicken CII. Arthritis was evaluated based on edema lesions in periarticular tissue and erythema of the four paws according to 0-4 grade of ascending severity as described elsewhere [18]: 0=normal; 1=mild erythema or swelling of a wrist or ankle or erythema and swelling of any severity for 1 digit; 2=more than three inflamed digits or moderate erythema and swelling of an ankle or wrist; 3=severe erythema and swelling inflammation of a wrist or ankle; 4=complete erythema and swelling of a wrist and ankle including all digits.
Histopathological arthritis assessments
The radiocarpal joints were dissected on day 45, fixed in 10% neutral-buffered formalin, decalcified in decalcifying solution (Wako Pure Chemicals, Osaka, Japan), and embedded in paraffin. The front paws were sectioned at 4 μm, deparaffinized with xylene, and hydrated with a graded alcohol series. The sections were stained with Hematoxylin and Eosin (H&E).
Preparation of anti-CII Fab fragments
DBA/1j mice were subcutaneously immunized with chicken CII and CFA into the base of the tail on day 0 and 21 according to the above described method. Blood samples were withdrawn by orbital sinus venipuncture on day 30. Serum was separated from clotting blood by centrifugation at 1500×g at 4°C for 15 minutes. The anti-CII pAbs were purified by Hitrap IgG protein an affinity chromatography (GE Healthcare Bio-Science, Sweden) and concentrated by Vivaspin-20 (Sartorius Stedium Biotech Gmbh, Germany) to 10 mg/ml in phosphatebuffered saline (PBS) based on an OD280. Fab fragments were prepared using 10 mg purified pAbs in 20 mM phosphate buffer containing 10 mM EDTA and 20 mM cysteine, co-incubated with 250 μg agaroselinked papain (Sigma Aldrich) at 37°C for 24 hours according to the methods described previously [13]. The Fab fragments were purified by Hitrap IgG protein an affinity chromatography. Absorbance at 280 nm was measured using a Gene Quant Pro spectrophotometer (GE Healthcare, UK) to determine the protein concentrations.
Serum collection and analysis
Blood samples were withdrawn by orbital sinus venipuncture on day 48. Serum was separated from clotting blood by centrifugation at 1500×g at 4°C for 15 minutes. The anti-CII IgG antibody was evaluated by direct Enzyme-Linked Immunosorbent Assay (ELISA) using plates coated with mouse CII. The complement component, including C3a protein, was measured by sandwich ELISA according to the manufacturer’s instructions for C3a ELISA kits (BD Biosciences, San Diego, CA, USA).
Competitive binding assay
The affinity of anti-CII pAbs to mouse CII inhibited by its Fab fragments was examined using ELISA. A mixture of anti-CII pAbs (50 μg/ml) and various concentrations of anti-CII Fab fragments (0-1,000 μg/ml) was prepared and incubated at 37°C for 1 hour in a 96-well plate coated with 1 μg/ml mouse CII. Phosphatase-conjugated antimouse IgG (Fc) (Sigma-Aldrich) was added and the color reaction was examined by p-nitrophenylphosphate substrate. Absorbance was measured at 405 nm by a microplate reader.
Complement inhibition assay
Fab fragment-inhibited complement activation was examined by ELISA with a modified procedure as described previously [19]. Briefly, 96-well plates were coated with 1 μg/ml mouse CII and incubated with the mixture of anti-CII pAbs (50 μg/ml) and various concentrations of Fab fragments (0-1,000 μg/ml) at 37°C for 1 hour. The mouse complement (Rockland Immunochemical, Gilbertsville, PA, USA) diluted in veronal saline buffer containing 1 mM MgCl2 and 2 mM CaCl2 was added and incubated at 37°C for 1 hour. The supernatants were separated to measure the level of C3a, and were measured by sandwich ELISA according to the manufacturer’s instructions for C3a ELISA kits (BD Biosciences).
Statistical analyses
The data are given as the means and Standard Error of the Mean (S.E.M.). The level of statistical significance was determined using the unpaired Student’s t-test for continuous variables and the Mann Whitney U-test for non-continuous variables. Values of p<0.05 were considered to indicate significance.
Effect of anti-CII Fabs on CIA in mice
To determine the efficacy of anti-CII Fabs, mice sensitized with chicken CII emulsified in CFA were given 1 mg anti-CII Fab fragments 24 hours prior to the first immunization of chicken CII. CIA developed rapidly in mice immunized with chicken CII and clinical signs first occurred within 3 days after the second chicken CII injection. The 80% and 100% incidence of CIA was observed by day 27 and 36, respectively. However, the 80% incidence of CIA in the anti-CII Fab-treated group appeared on day 41 and the 100% incidence was observed on day 43 (Figure 1A). Furthermore, the mean maximum arthritis score of the control Fabs-treated group was reached between day 43 (10.65) and day 48 (10.11), whereas the anti-CII Fabs-treated group showed the protection of severe joint inflammation throughout the observation period (mean arthritis score<6) (Figure 1B). On the other hand, although the weight of control Fabs-treated mice gradually declined after the occurrence of clinical arthritis, anti-CII Fabs significantly conserved the loss of body weight from the onset of CIA until termination (Figure 1C).
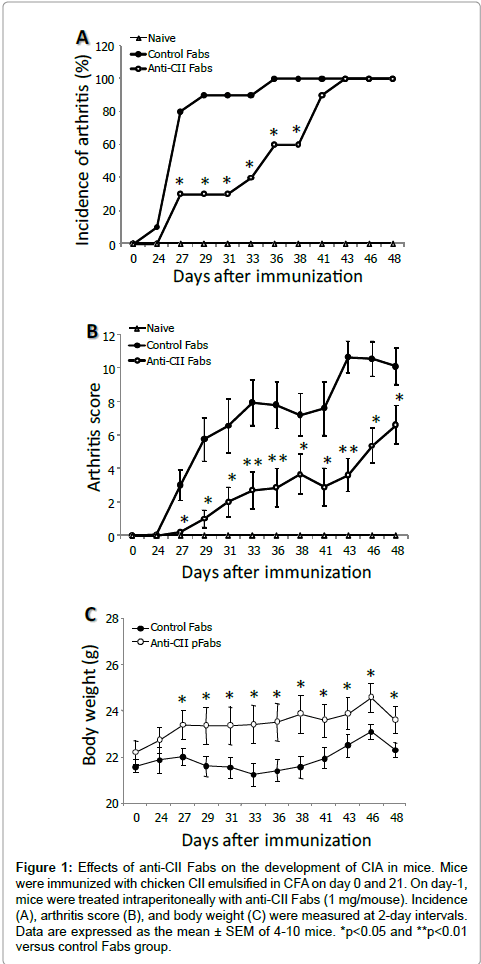
Figure 1: Effects of anti-CII Fabs on the development of CIA in mice. Mice were immunized with chicken CII emulsified in CFA on day 0 and 21. On day-1, mice were treated intraperitoneally with anti-CII Fabs (1 mg/mouse). Incidence (A), arthritis score (B), and body weight (C) were measured at 2-day intervals. Data are expressed as the mean ± SEM of 4-10 mice. *p< 0.05 and **p< 0.01 versus control Fabs group.
Effect of anti-CII Fab on histopathological changes in CIA mice
The histopathological examination of front paws was performed on day 48 after the first immunization. Representative H&E staining of naïve control Fabs-treated, and anti-CII Fabs-treated mice are shown in Figures 2A-2C, respectively. Histological features of the radiocarpal joints in control Fabs-treated mice revealed the numerous accumulations of mononuclear cells and polymorphonuclear leukocytes in the synovial tissues. Cartilage destruction and bone erosion were slightly evidenced; however, hyperplasia and hypertrophy of synoviocytes, synovitis and pannus formation were quite prominent. In contrast, the animals given anti-CII Fabs had reduced leukocytic infiltration with a thin layer of the synovial membrane and no joint destruction.
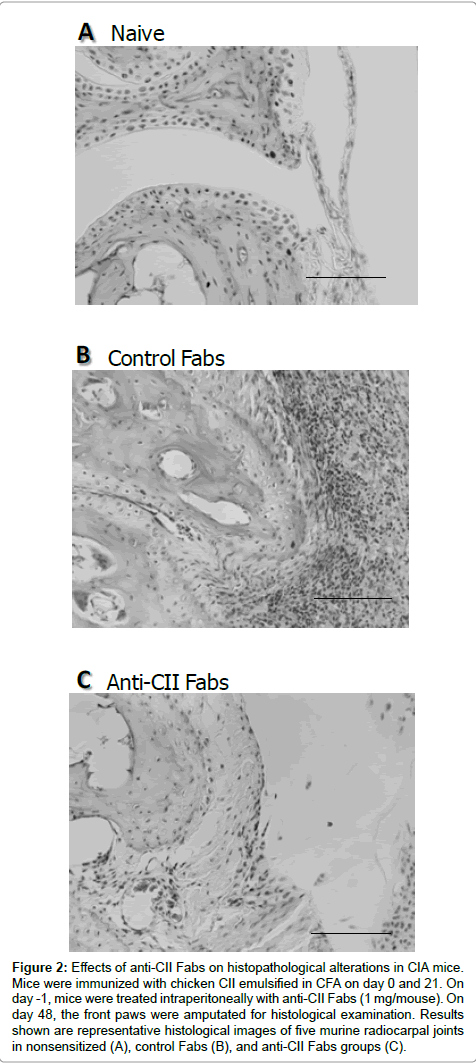
Figure 2: Effects of anti-CII Fabs on histopathological alterations in CIA mice. Mice were immunized with chicken CII emulsified in CFA on day 0 and 21. On day -1, mice were treated intraperitoneally with anti-CII Fabs (1 mg/mouse). On day 48, the front paws were amputated for histological examination. Results shown are representative histological images of five murine radiocarpal joints in nonsensitized (A), control Fabs (B), and anti-CII Fabs groups (C).
Effect of anti-CII Fabs on production of anti-CII IgG antibody in serum of CIA mice
To compare the humoral immune response against CII between control Fabs-treated and anti-CII Fabs-treated groups, the level of serum anti-CII IgG antibody was measured on day 48 after the first immunization of chicken CII. The results demonstrated that the anti- CII IgG of both groups was significantly elevated compared to that of normal mice; however, formation in the anti-CII Fab-treated group was not disrupted by the administration of Fab fragments (Figure 3A).
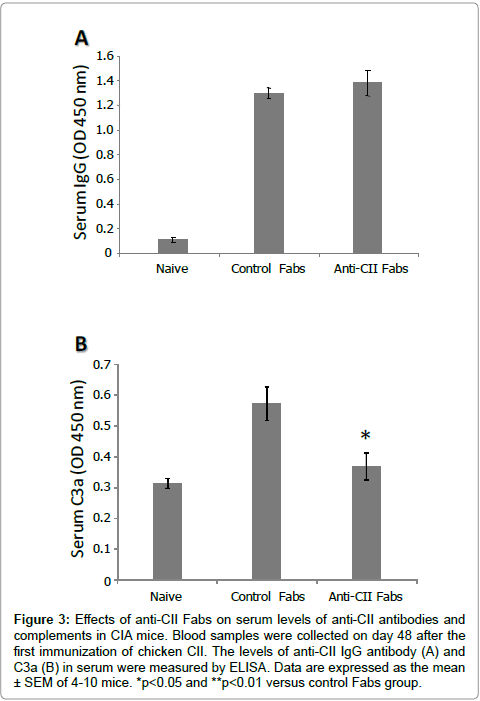
Figure 3: Effects of anti-CII Fabs on serum levels of anti-CII antibodies and complements in CIA mice. Blood samples were collected on day 48 after the first immunization of chicken CII. The levels of anti-CII IgG antibody (A) and C3a (B) in serum were measured by ELISA. Data are expressed as the mean ± SEM of 4-10 mice. *p< 0.05 and **p< 0.01 versus control Fabs group.
Effect of anti-CII Fabs on concentration of complements in serum of CIA mice. The serum level of the C3a component was evaluated to clarify the in vivo effect of anti-CII Fabs on the activity of the classical complement pathway. The result demonstrated that the serum C3a level of control Fabs-treated mice was higher than that of normal mice, whereas anti-CII Fabs significantly inhibited its production (Figure 3B).
Effect of anti-CII Fabs on interaction of mouse CII and anti- CII pAbs
To examine whether the function of anti-CII Fabs has potential for competitive binding to block the interaction of mouse CII and anti-CII pAbs, anti-CII pAbs alone or the mixture of anti-CII pAbs and various concentrations of anti-CII Fabs was incubated in an ELISA plate coated with mouse CII. The result showed that the affinity of anti-CII pAbs fell by anti-CII Fabs in a concentration-dependent manner (Figure 4A). The binding of anti-CII pAbs was significantly suppressed by anti-CII pFabs at concentrations of 100 and 1000 μg/ml, with reduction rates of 17% and 40%, respectively.
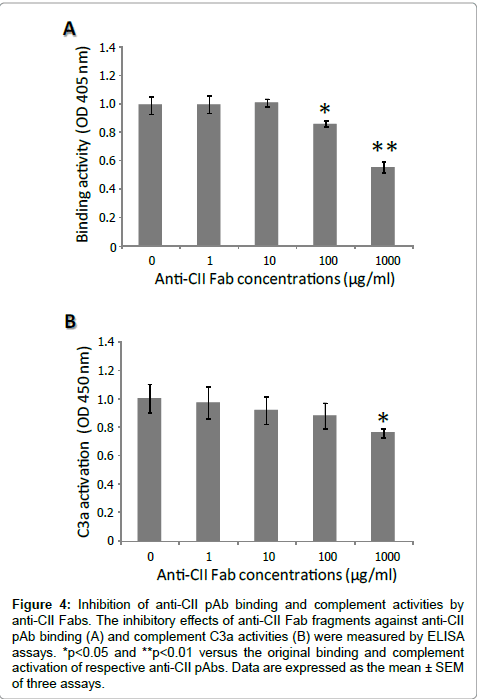
Figure 4: Inhibition of anti-CII pAb binding and complement activities by anti-CII Fabs. The inhibitory effects of anti-CII Fab fragments against anti-CII pAb binding (A) and complement C3a activities (B) were measured by ELISA assays. *p< 0.05 and **p< 0.01 versus the original binding and complement activation of respective anti-CII pAbs. Data are expressed as the mean ± SEM of three assays.
Effect of anti-CII Fabs on complement activation
To investigate the effect of anti-CII Fabs on complement activation, anti-CII pAbs alone or the mixture of anti-CII pAbs and various concentrations of anti-CII pFab fragments was incubated in an ELISA plate coated with mouse CII. Figure 4B showed that the C3a level in the supernatant was gradually reduced by anti-CII Fabs in a concentration-dependent manner. The cleavage product C3a was significantly deceased at the highest concentration of 1000 μg/ml, with a reduction rate of 24%.
Our previous study demonstrated that antibody-mediated inflammatory disease was able to be specifically regulated by Fabs of the mediating antibodies, as AOA-MA in mice was markedly suppressed by anti-OVA Fabs [13]. However, whether CIA in mice, which resembles human RA, was specifically regulated by anti-CII Fabs remained unclear. In this study, we extended our previous work to assess the inhibitory effect of anti-CII Fabs on CIA in mice; as a result, treatment with anti-CII Fabs suppressed CIA in mice. Current therapeutic drugs for RA include steroidal and non-steroidal antiinflammatory drugs, immunosuppressive agents, disease-modifying anti-rheumatic drugs such as methotrexate, and biological agents including infliximab [14-16]. However, no antigen-specific drug therapy for RA is clinically available currently. Thus, our findings suggest that RA may be specifically treated with antigen-specific Fabs of the mediating antibodies.
Because it is widely recognized that the antibody-induced arthritis model skips the phase of B cell contribution [20], this allowed us to study the effects of anti-CII Fabs on CIA in mice. Furthermore, CIA is proposed to be T-cell antigen-specific [21], which is based principally on its immunological and pathological similarities to human RA; therefore, this model has been widely used to evaluate the therapeutic efficacies of drug candidates for RA [22,23]. In the present study, the incidence and severity of CIA were markedly suppressed by administration of anti-CII Fabs. Furthermore, the prevention of autoimmune diseases has been reported by using antibody fragments, including autoimmune hemolytic diseases [24] and bullous pemphigoid [12]. These data confirm that antigen-specific suppression of autoimmunity is simply achieved by employing Fabs of the mediating antibodies.
The IC formation of arthricular CII and rheumatoid factors including IgG is presumably triggered in the initial step of the progressively immunopathological response of RA [25]. In addition, the potential of anti-CII Fabs against the pathogenesis of RA was determined by using an in vitro competitive CII binding assay. In this study, we showed that Fab fragments competitively inhibited the affinity of anti-CII pAbs to murine CII, because much greater absorbance of whole anti-CII antibodies was observed when the CII coating was incubated with anti-CII pAbs alone than with the cocktail of anti-CII pAbs and anti-CII Fabs. Moreover, the in vivo experiment demonstrated that both the incidence and severity of CIA mice treated with the Fabs were markedly decreased, although autoantibodies were still detectable. Thus, our studies indicated that the specific regulation of CIA by anti-CII Fabs is not associated with the reduced production of anti-CII IgG antibody, but CIA mice treated with antigen-specific Fabs were protected from pathogenic autoantibodies binding to the corresponding arthricular CII autoantigen.
We also observed that the consumption of serum C3 was reduced in CIA mice treated with anti-CII Fabs, which lacks the Fc portion as the binding site of complements; furthermore, the in vitro complement inhibition assay showed that ICs-mediated activation of C3 complement was suppressed by anti-CII Fabs. It has been reported that the provoked level of C3a in the serum of RA patients is correlated with laboratory and clinical manifestations of RA [4,26], whereas the reduced levels of C3 are comparable to those of healthy subjects [27]. The activated protein of C3a is a potent mediator involved in inflammatory responses, including alteration of vascular permeability, opsonization for inflammatory leukocytes and activation of release pro-inflammatory cytokines [28]; moreover, murine CIA suppressed by the C3 convertase inhibitor has been reported [29]. These previous findings strongly indicated that the development of antibody-mediated disease results from IC-activated complement [30,31]. Thus, these data suggest that the prevention of CII-bound autoantibodies by anti-CII Fabs appears to be the mechanism by which the pathogenesis of CIA was suppressed via escape from complement recognition.
It has been reported that no clinical development in FcγR-/- mice immunized with CII was observed [32]. Furthermore, the Fab fragments lacked the site for FcγRs that play crucial roles in the development of arthritis [33]. In this study, we confirmed that the accumulation of FcγR-bearing leukocytes, including neutrophils and macrophages, in synovial tissues was markedly suppressed by anti-CII Fab. Moreover, it has been documented that bone destruction in CIA mice results from tissue-degrading enzymes released by neutrophils and macrophages [34], suggesting that the normal articular cartilage and bone in the anti-CII Fabs-treated group are associated with protection from the infiltrating inflammatory cells. In conclusion, the use of chemical agents in symptomatic treatment has shown limited efficacy and the risk of adverse drug reactions; therefore, antigen-specific therapy is a promising alternative attractive remedy for routine use in RA patients. Unfortunately, pathogenic antigen-specific therapy is clinically unavailable. In the present study, we successfully prepared Fabs from anti-CII antibodies to inhibit RA development. This inhibitory effect on the pathogenesis of RA is associated with interference with antigen–antibody interactions; therefore, the activities of the complement pathway and inflammatory cells are down-regulated. These findings identified antigen-specific Fabs of the mediating antibodies as a new therapeutic strategy for the treatment of RA.
